논문 정보
- 제목: Universal Cell Embeddings: A Foundation Model for Cell Biology
- 저자: Yanay Rosen, Yusuf Roohani 외
- 형태: bioRxiv preprint (업로드된 PDF 기준, peer review 전 버전)
- 핵심 질문: 서로 다른 조직, 서로 다른 실험 배치, 서로 다른 종에서 나온 single-cell RNA-seq 데이터를 재학습 없이 하나의 세포 표현 공간으로 보낼 수 있을까?
3줄 요약
UCE는 single-cell RNA-seq 데이터를 위한 foundation model로, 각 세포를 하나의 universal embedding으로 바꾼다.
핵심은 유전자를 단순한 gene ID가 아니라 단백질 서열 기반 의미 표현(protein embedding) 으로 다루어, 새로운 종에도 zero-shot으로 확장된다는 점이다.
이 논문은 UCE가 36M cells, 300개 이상 dataset, 8개 species를 하나의 공간으로 통합하고, 새로운 데이터와 새로운 종, 심지어 새로운 biological question에도 추가 fine-tuning 없이 적용될 수 있음을 보여준다.
숫자로 보는 이 논문
- 33-layer transformer
- 650M parameters
- 1280차원 cell embedding
- 36 million cells
- 300개 이상 dataset
- 50 tissues
- 8 species
- 1,000개 이상 uniquely named cell types
- 24개 A100 80GB GPU에서 40일 학습
이 숫자만 봐도 저자들이 UCE를 단순한 “분석 기법”이 아니라, 재사용 가능한 세포 표현 인프라로 설계했다는 점이 드러난다.
왜 중요한가
single-cell RNA-seq 분석의 큰 병목은 늘 비슷했다.
새 데이터셋이 들어오면 다시 정규화하고, 다시 integration하고, 다시 label transfer를 하고, 다시 classifier를 학습해야 한다.
특히 서로 다른 실험 조건, 플랫폼, batch, tissue, species가 섞이면 batch effect와 feature mismatch(측정된 gene set 차이) 때문에 기존 표현 공간이 쉽게 무너진다.
이 논문이 겨냥한 문제는 명확하다.
“세포를 표현하는 좌표계 자체를 보편적으로 만들 수 있는가?”
즉, 새로운 데이터가 들어와도 같은 좌표계 위에 바로 올릴 수 있는 표현 공간을 만들자는 것이다.
논문에서 UCE의 진짜 차별점은 성능 수치 자체보다, 새 데이터가 들어와도 representation space를 바꾸지 않는다는 점이다.
이게 가능해지면 atlas는 정적인 참고자료가 아니라, 새로운 세포를 즉시 대조해 볼 수 있는 검색 가능한 reference space가 된다.
UCE의 핵심 아이디어
1) Cell을 “bag of RNA”로 본다
저자들은 세포를 길게 정렬된 유전자 문장으로 보기보다, 우선 발현된 유전자들의 가중 샘플로 본다.
각 세포에서 발현된 gene만, 발현량에 비례하도록 1024개를 중복 허용 샘플링한다.
이 설계는 두 가지 점에서 중요하다.
첫째, scRNA-seq의 sparsity를 어느 정도 흡수한다.
둘째, 어떤 dataset는 특정 gene이 없고 다른 dataset는 있는 상황에서도, 고정된 gene vocabulary에 덜 묶인다.
2) Gene token을 species-specific ID가 아니라 protein embedding으로 표현한다
여기가 이 논문의 가장 흥미로운 포인트다.
UCE는 gene 자체를 바로 token으로 쓰지 않고, 그 gene이 coding하는 protein의 embedding을 쓴다.
이 protein embedding은 ESM2에서 가져온다.
즉, UCE는 “이 gene 이름이 무엇이냐”보다 “이 gene이 어떤 단백질적 의미를 가지느냐”에 더 가까운 표현을 사용한다.
그래서 훈련에 없던 species라도, 그 종의 protein-coding gene 서열만 있으면 같은 의미 공간 위에 매핑할 가능성이 생긴다.
3) Chromosome 정보도 함께 넣는다
샘플링된 gene들은 chromosome별로 묶고, genomic location 기준으로 정렬한 뒤 transformer에 넣는다.
다만 저자들 스스로도 Methods에서 언급하듯, 이 ordering은 성능에 오히려 약간 불리하게 작용했다.
즉, biologically motivated design이긴 하지만, 이 부분은 앞으로 바뀔 여지가 있는 설계다.
4) Self-supervised objective로 학습한다
UCE는 cell type label을 쓰지 않는다.
훈련 시 발현된 gene의 일부를 masking한 뒤, “이 gene이 이 cell에서 발현되었는가?” 를 맞히도록 학습한다.
다시 말해, 세포 전체 embedding과 gene embedding을 결합해 binary expression prediction을 수행하는 self-supervised 방식이다.
이 선택의 의미는 분명하다.
저자들은 사람이 붙인 cell type label이 편향되거나 해상도가 들쑥날쑥할 수 있다고 보고, 세포 공간의 구조가 label 없이 emergence 하도록 두고자 했다.
Figure 1. UCE는 어떻게 세포를 universal space로 보낼까

원 논문 Figure 1a에서 핵심 패널만 crop.
이 그림의 메시지는 단순하다.
“RNA counts → gene sampling → protein embedding → transformer → cell embedding” 이다.
여기서 꼭 짚어야 할 핵심은 세 가지다.
- 입력은 single-cell RNA expression이지만, 내부 token은 protein-level representation이다.
- 학습 목표는 cell type 분류가 아니라 masked gene expression prediction이다.
- 결과적으로 얻고 싶은 것은 task-specific classifier가 아니라 범용 cell embedding이다.
핵심 해석
UCE는 gene expression을 “텍스트처럼 읽는 모델”이라기보다, 세포를 단백질 의미 공간 위에서 재표현하는 모델에 가깝다.
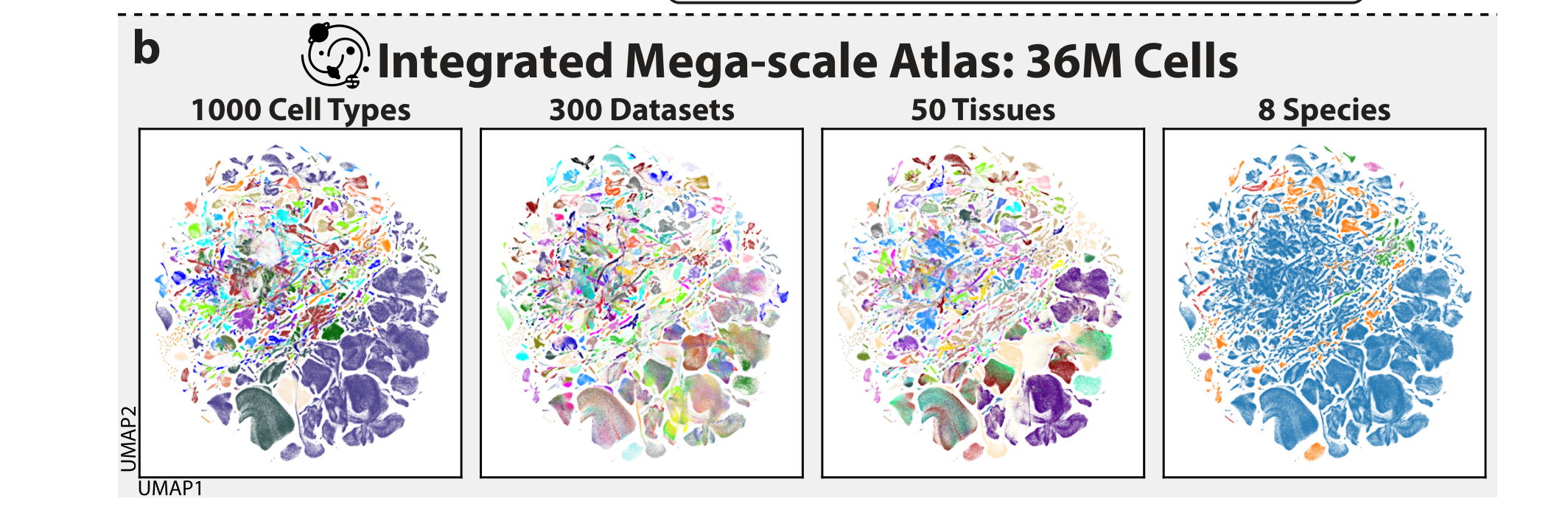
원 논문 Figure 1b에서 핵심 패널만 crop.
Figure 1b는 UCE가 만든 Integrated Mega-scale Atlas(IMA) 를 보여준다.
저자들은 36M cells를 하나의 embedding space에 올렸고, 그 공간 안에서 cell type, dataset, tissue, species 수준의 구조를 동시에 살펴본다.
이 그림에서 봐야 할 점은 “많이 모았다”가 아니다.
더 중요한 메시지는 세포 타입은 모이고, batch 같은 실험 조건은 덜 전면에 드러나는 공간을 만들었다는 것이다.
이 논문에서 가장 중요한 주장: zero-shot이어야 진짜 universal하다
저자들이 반복해서 강조하는 문장은 사실상 이것이다.
새 데이터에 맞춰 model을 fine-tune하면, representation space 자체가 바뀌기 때문에 더 이상 universal하다고 보기 어렵다.
즉, UCE는 “좋은 임베딩”보다 한 걸음 더 나가서, 새 데이터가 와도 같은 좌표계가 유지되는가를 중심 가치로 둔다.
Figure 2. 새로운 데이터와 새로운 종에도 바로 적용되는가

원 논문 Figure 2a-b에서 핵심 패널만 crop.
Figure 2a는 개념도이고, Figure 2b는 실제 결과다.
개념적으로 zero-shot model은 새 데이터를 기존 representation space에 바로 매핑한다.
반면 fine-tuned model은 새 데이터에 맞게 model이 업데이트되고, 그 결과 공간 자체가 달라진다.
논문은 이 주장을 Tabula Sapiens v2로 테스트한다.
Tabula Sapiens v2는 581,430 cells, 27 tissues, 167 batches, 162 unique cell types로 구성된 큰 human dataset이다.
저자들에 따르면 UCE는 이 unseen dataset에 대해, zero-shot setting에서 Geneformer 대비 전체 점수 13.9%, biological conservation 16.2%, batch correction 10.1% 더 좋았다.
더 나아가 저자들은 dataset-specific training이 필요한 scVI, scArches보다도 약간 더 낫다고 주장한다.
이 결과가 중요한 이유는 단순한 SOTA 경쟁이 아니다.
핵심은 “재학습하지 않았는데도 성능이 유지된다” 는 점이다.
이게 성립하면 atlas는 매번 다시 맞추는 reference가 아니라, 바로 연결되는 공통 좌표계가 된다.

원 논문 Figure 2c-d에서 핵심 패널만 crop.
Figure 2c-d는 species generalization을 보여준다.
저자들은 훈련에 없던 green monkey dataset을 넣었고, lymph node cell type 구조가 자연스럽게 나타나는 것을 보인다.
그 다음 human lymph node embedding에 학습된 간단한 classifier를 green monkey에 적용했을 때, 예측 label이 원 주석과 높은 합의를 보인다고 보고한다.
여기서 포인트는 “cross-species label transfer가 된다”가 아니다.
더 중요한 메시지는 ortholog mapping을 강하게 전제하지 않고도, 단백질 의미 공간을 통해 새로운 종을 바로 같은 임베딩 공간에 놓을 수 있다는 것이다.
다만 이 논문은 동시에 중요한 한계도 인정한다.
fly(Tabula Drosophilae) 에서는 예측이 부정확했다.
즉, UCE는 “모든 종에 대해 완전히 보편적”이라기보다, 훈련 종들과 너무 멀지 않은 종들에서는 매우 강력하지만, 진화적으로 먼 종에서는 약해질 수 있는 모델이다.
핵심 해석
UCE의 universality는 절대적인 의미의 보편성이라기보다, protein meaning space를 매개로 한 practical universality에 가깝다.
Figure 3. UCE 공간은 정말 biological meaning을 갖고 있는가

원 논문 Figure 3a-b에서 핵심 패널만 crop.
Figure 3a는 unseen lung dataset을 UCE 공간에 올렸을 때 cell type dendrogram이 어떻게 조직되는지 보여준다.
T cell, monocyte, endothelial cell 같은 개별 타입뿐 아니라, immune / epithelial 같은 상위 범주 구조도 드러난다.
Figure 3b는 더 흥미롭다.
저자들은 UCE embedding에서 cell-cell distance와 Cell Ontology tree distance를 비교했고, ontology상 더 멀리 떨어진 cell type일수록 embedding distance도 커진다고 보였다.
이건 단순히 “클러스터가 잘 나뉜다”는 얘기가 아니다.
UCE 공간이 세포 타입 간 상대적 관계까지 어느 정도 보존한다는 주장이다.
논문에는 이런 emergent biological structure에 대한 추가 증거도 나온다.
- mesoderm / endoderm / ectoderm 같은 germ layer 기원이 공간에서 함께 모인다.
- held-out cell type의 germ layer를 예측하는 classifier 정확도가 80% 이상이다.
- bone marrow 같은 unseen data에서도 발달 단계와 lineage 구조가 반영된다.
즉, UCE는 단순 annotation tool보다 developmental geometry를 어느 정도 내재화한 representation을 만들려는 시도에 가깝다.
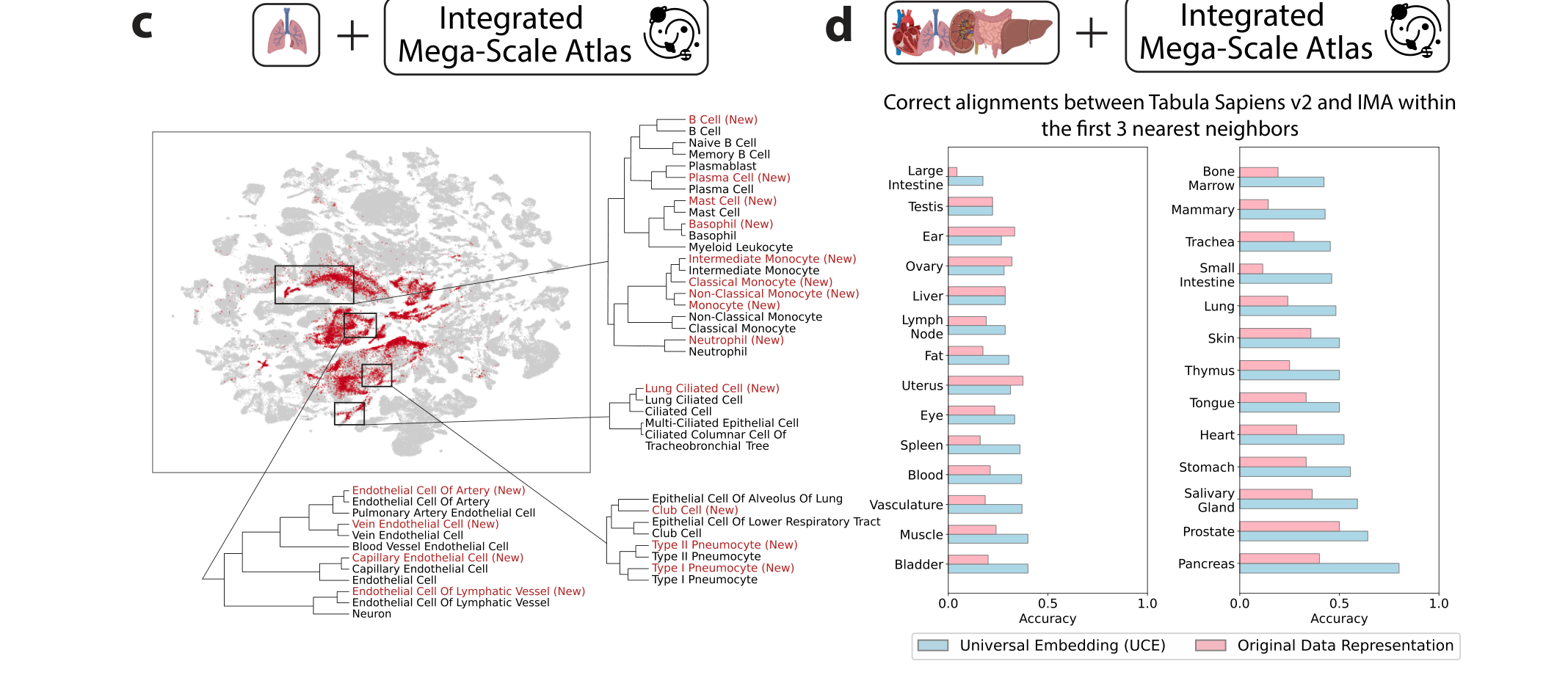
원 논문 Figure 3c-d에서 핵심 패널만 crop.
Figure 3c-d는 unseen dataset을 IMA에 직접 붙였을 때 얼마나 잘 align되는지 보여준다.
저자들은 Tabula Sapiens v2와 IMA 사이의 cell type centroid를 비교했고, 3-nearest neighbor 기준 alignment 정확도가 원래 gene expression space보다 65% 더 높다고 보고한다.
single nearest centroid 기준으로는 92% 개선이라고 설명한다.
여기서 중요한 포인트는, raw expression space에서의 단순 거리보다 UCE space에서의 거리가 훨씬 더 biological meaning을 가진다는 점이다.
즉, 이 임베딩 공간은 단순 압축이 아니라 reference matching을 위한 작동하는 좌표계가 된다.
Figure 4. UCE는 “분석 도구”를 넘어 “가설 생성 엔진”이 될 수 있을까

원 논문 Figure 4a-b에서 핵심 패널만 crop.
Figure 4는 이 논문의 응용 파트를 대표한다.
저자들은 mouse kidney의 Norn cell을 사례로 든다.
Norn cell은 kidney에서 erythropoietin(Epo)을 만드는 것으로 알려진 fibroblast-like cell이다.
저자들의 workflow는 세 단계다.
- 새 cell type cluster를 발견한다.
- 같은 tissue의 다른 dataset에서 그 cell type을 다시 찾는다.
- 더 나아가 다른 tissue 전체에서 비슷한 상태의 세포를 찾는다.
즉, UCE는 단순 integration을 넘어서, “이 세포와 비슷한 상태를 가진 세포가 생물학 전체에서 어디 더 있는가?” 를 묻는 도구로 쓰인다.

원 논문 Figure 4c-e에서 핵심 패널만 crop.
저자들은 mouse kidney embedding에 학습된 간단한 logistic classifier를 IMA 전체에 적용해 Norn-like cell을 찾는다.
그 결과 kidney뿐 아니라 lung, heart 등 다른 tissue에서도 Norn marker와 유사한 발현 패턴을 가진 세포가 관찰된다고 주장한다.
더 나아가 lung disease dataset(IPF, COPD, control)에서도 Norn-like cell을 보고, 질환군별로 marker gene expression이 다르다고 해석한다.
특히 COPD와 IPF는 모두 혈중 Epo 증가와 관련이 있지만 양상이 다르고, 이 차이가 Norn-like cell state와 연결될 수 있다는 가설을 제시한다.
이 사례는 논문의 방향성을 잘 보여준다.
UCE의 목적은 단순히 “cell type을 잘 맞히는 것”이 아니라, 아직 이름 붙지 않았거나 흩어져 있는 생물학적 유사성을 찾는 것이다.
핵심 해석
Figure 4는 UCE를 atlas integration model이 아니라, unbiased search engine for cell biology로 보게 만든다.
이 논문이 정말 새롭게 만든 것
이 논문의 기여는 세 가지 층위에서 정리할 수 있다.
1) 기술적 기여
single-cell foundation model에 protein language model 기반 gene representation을 본격적으로 연결했다.
이 덕분에 fixed gene vocabulary나 species-specific homolog mapping에 덜 의존한다.
2) 방법론적 기여
새 데이터에 맞춰 model을 바꾸지 않는 zero-shot universal embedding을 전면에 내세웠다.
이건 기존 single-cell integration이 자주 빠지는 “새 dataset마다 다시 학습” 루프를 끊으려는 시도다.
3) 개념적 기여
atlas를 static resource가 아니라, 새로운 세포를 즉시 투영하고 비교할 수 있는 공통 좌표계로 본다.
이 관점은 annotation, transfer learning, hypothesis generation을 하나의 공간 문제로 묶는다.
강점
1. “Universal”이라는 말을 비교적 엄격하게 사용한다
새 dataset에 맞춰 fine-tune하지 않는다는 기준을 명확히 세웠다.
그래서 “generalizable하다”는 말이 비교적 실질적이다.
2. Species 확장성의 설계 논리가 설득력 있다
gene ID가 아니라 protein embedding을 쓰기 때문에, 새로운 species를 같은 공간에 넣는 이유가 명확하다.
3. Label-free training 철학이 일관적이다
cell type label을 학습에 직접 쓰지 않으면서도, 결과 공간에서 cell type / lineage / ontology 구조가 emergence 한다는 점은 이 논문의 가장 강한 메시지다.
4. 실제 biological use case가 있다
Norn cell 사례 덕분에 이 논문은 “벤치마크 점수 좋은 모델”에서 끝나지 않는다.
실제로 새로운 biological 연결고리를 찾는 도구로 보인다.
한계와 비판적으로 볼 점
1. 아직 preprint다
업로드된 PDF 기준 이 논문은 bioRxiv preprint이며 peer review 전이다.
핵심 아이디어는 강하지만, 일부 해석과 benchmark framing은 후속 검증이 더 필요하다.
2. 모든 종에 대해 보편적이지는 않다
green monkey, naked mole rat, chicken에서는 좋은 결과가 나오지만, fly에서는 실패한다.
즉 “universal”은 현실적으로 훈련 분포 주변에서 강한 보편성으로 읽는 편이 정확하다.
3. transcript-level biology를 담지 못한다
저자들도 Discussion에서 인정하듯, 현재 UCE는 raw RNA transcript, splicing, variant, non-coding RNA 정보를 거의 쓰지 않는다.
그래서 세포 생물학의 중요한 축 일부는 여전히 빠져 있다.
4. transformer context limit의 제약이 있다
한 세포당 1024 gene 샘플만 넣기 때문에, 세포 내 모든 정보를 완전히 쓰는 구조는 아니다.
이는 quadratic attention cost 때문에 생긴 현실적 타협이다.
5. chromosome ordering은 biologically motivated이지만 성능상 이득이 크지 않다
Methods에서는 이 ordering이 오히려 moderate negative effect가 있었다고 적는다.
즉, 모델 설계의 일부는 아직 “좋은 아이디어” 단계이지, 완전히 검증된 선택은 아니다.
6. biological truth의 평가가 여전히 label과 ontology에 의존한다
이 논문도 결국 기존 annotation과 Cell Ontology를 많이 참조한다.
즉, foundation model의 진짜 가치를 평가하는 benchmark는 아직 충분히 정립되지 않았다.
강조 포인트
포인트 1. “좋은 임베딩”이 아니라 “안 바뀌는 좌표계”가 핵심
이 논문은 성능 숫자보다 representation space를 유지하는 것을 더 본질적인 가치로 둔다.
블로그에서 이 부분을 앞에 두면 논문의 차별점이 훨씬 명확해진다.
포인트 2. 유전자를 텍스트가 아니라 단백질 의미 공간으로 옮겼다
single-cell foundation model 중에서도 UCE의 가장 독특한 부분은 protein LM을 gene representation의 기반으로 쓴 점이다.
이 덕분에 cross-species zero-shot이 단순 trick이 아니라 설계 철학이 된다.
포인트 3. 이 모델의 최종 목적은 annotation 자동화가 아니다
Figure 4가 보여주듯, UCE의 지향점은 새로운 cell state와 function을 검색하는 것이다.
즉, 이 모델의 가장 흥미로운 사용처는 “이 세포 이름이 뭐지?”보다 “이 세포와 닮은 biology가 어디 더 있지?” 에 가깝다.
이 논문을 한 문장으로 정리하면
UCE는 single-cell 데이터를 위한 “범용 좌표계”를 만들려는 시도이며, 그 핵심은 protein meaning space를 이용해 세포를 zero-shot으로 같은 공간에 매핑한다는 점이다.
마무리
이 논문이 흥미로운 이유는 단지 규모가 커서가 아니다.
UCE는 single-cell analysis를 “새 dataset마다 다시 학습하는 문제”에서 “이미 만들어진 universal space에 새 세포를 놓는 문제”로 바꾸려 한다.
만약 이 방향이 더 발전한다면, 앞으로의 cell atlas는 정적인 reference가 아니라, 새 세포 상태를 즉시 해석하고 새로운 biological 가설을 끌어내는 검색 가능한 생물학적 좌표계가 될 수 있다.
참고 메모
- 본 문서는 업로드된 PDF 논문을 바탕으로 작성했다.
- Figures는 원 논문 Figure 1–4 (pp. 28–31) 에서 캡션과 페이지 번호를 제외하고 핵심 패널 중심으로 crop했다.
- 이해를 돕기 위해 일부 설명은 블로그용 문체로 재구성했다.
- 원 논문은 preprint이므로, 후속 peer review 및 후속 연구와 함께 보는 것이 좋다.
'AI 생성 글 정리 > bio' 카테고리의 다른 글
| 논문 정리: Benchmarking DNA Foundation Models: Biological Blind Spots in Evo2 Variant-Effect Prediction (0) | 2026.04.03 |
|---|---|
| PATH-ORACLE 논문 핵심 정리 (0) | 2026.04.03 |
| 논문 정리: Benchmarking zero-shot single-cell foundation model embeddings for cellular dynamics reconstruction (0) | 2026.04.02 |
| Geneformer 논문 핵심 정리 (0) | 2026.04.02 |
| HyenaDNA 논문 핵심 정리 (0) | 2026.04.02 |



