논문: Chai Discovery team, Chai-1: Decoding the molecular interactions of life
형태: bioRxiv preprint (peer review 전)
게시 버전: 2024-10-11 posted
DOI: https://doi.org/10.1101/2024.10.10.615955
비고: 이 글의 숫자와 해석은 업로드된 원문 PDF를 기준으로 정리했다.
한눈에 보는 결론
Chai-1은 단백질, DNA/RNA, small molecule까지 함께 다루는 멀티모달 구조 예측 foundation model이다. 이 논문의 핵심은 단순히 “정확도가 높다”에 그치지 않는다. 더 중요한 포인트는 세 가지다.
- 실험 제약조건을 프롬프트처럼 넣을 수 있다.
Epitope mapping, cross-link mass spectrometry, pocket/contact restraints 같은 wet-lab 정보를 구조 예측에 직접 반영할 수 있다. - MSA 없이도(single-sequence) 실용적인 성능을 낸다.
특히 multimer와 antibody 관련 작업에서 “MSA가 꼭 있어야만 한다”는 전제를 꽤 많이 무너뜨린다. - 공개 접근성이 좋다.
논문 기준으로는 모델 가중치와 추론 코드는 non-commercial 용도로 공개되고, 웹 인터페이스는 commercial drug discovery까지 사용할 수 있도록 제공된다고 설명한다.
한 문장으로 줄이면 이렇다.
Chai-1은 AlphaFold3 계열의 biomolecular structure prediction 흐름을 공개형·제약조건형·single-sequence 친화형으로 확장한 모델이다.
왜 이 논문이 중요한가
구조 예측 모델은 이미 “단백질 한 개를 접는 문제”를 넘어섰다. 실제 drug discovery에서는 다음이 더 중요하다.
- 단백질과 리간드가 어떻게 붙는지
- 단백질-단백질 인터페이스가 어떻게 형성되는지
- 항체-항원처럼 복잡하고 가변적인 상호작용을 어떻게 다룰지
- 실험에서 얻은 불완전한 정보(epitope, cross-link, pocket 정보)를 예측에 어떻게 섞을지
Chai-1은 바로 이 실무형 질문을 겨냥한다. 그래서 이 논문은 “구조 예측 성능 보고서”라기보다, 실험과 계산을 연결하는 분자 구조 foundation model의 방향을 보여주는 논문으로 읽는 편이 좋다.
모델은 어떻게 구성되어 있나

원문 Figure 1. Chai-1은 서열, 템플릿, MSA, language model embedding, wet-lab 제약조건, small molecule 정보를 함께 받아 구조를 예측한다.
논문에 따르면 Chai-1의 기본적인 아키텍처와 학습 전략은 AlphaFold 3 계열 설계를 상당 부분 따른다. 다만 몇 가지 차별점이 있다.
1) language model embedding 추가
기존 구조 예측 모델은 MSA에 크게 의존한다. Chai-1은 여기에 protein language model embedding을 추가해서 single-sequence 모드에서도 꽤 강한 성능을 낼 수 있게 만들었다.
2) constraint feature를 정식 입력으로 취급
Chai-1의 진짜 차별점은 여기다. 논문은 세 가지 제약 입력을 강조한다.
- Pocket constraints
- Contact constraints
- Docking constraints
이 정보는 템플릿처럼 “구조 힌트”를 주지만, 특히 inter-chain distance를 알려줄 수 있다는 점에서 의미가 크다. 실제 실험에서 얻은 epitope 정보나 cross-link 정보가 여기에 해당한다.
3) small molecule / modified residue 처리
리간드와 변형 잔기(modified residues)를 별도 고려하고, conformer generation 흐름도 포함한다.
이 점은 나중에 장점이자 한계로 다시 등장한다.
핵심 포인트
Chai-1의 핵심은 “AF3류 구조 예측 모델을 공개형으로 재현했다”가 아니라, 실험 제약을 받아들이는 모델 인터페이스를 만들었다는 데 있다.
결과를 읽기 전에: 지표를 아주 짧게 정리
논문에 등장하는 숫자는 지표가 섞여 있어서, 먼저 읽는 법을 잡아두면 편하다.
- Ligand PoseBusters: pocket-aligned ligand RMSD < 2 Å 이면 성공
- Protein-protein / antibody-protein: DockQ > 0.23 이면 acceptable 성공
- Protein monomer: Cα-LDDT가 높을수록 좋음
- Confidence: ipTM이 높을수록 인터페이스 품질이 좋다는 뜻
즉, 이 논문은 태스크별로 다른 지표를 쓰고 있으므로, 숫자 자체보다 어떤 문제에서 누구를 이겼는지를 읽는 것이 중요하다.
가장 중요한 결과 1: 단백질-리간드에서 AF3와 거의 동급
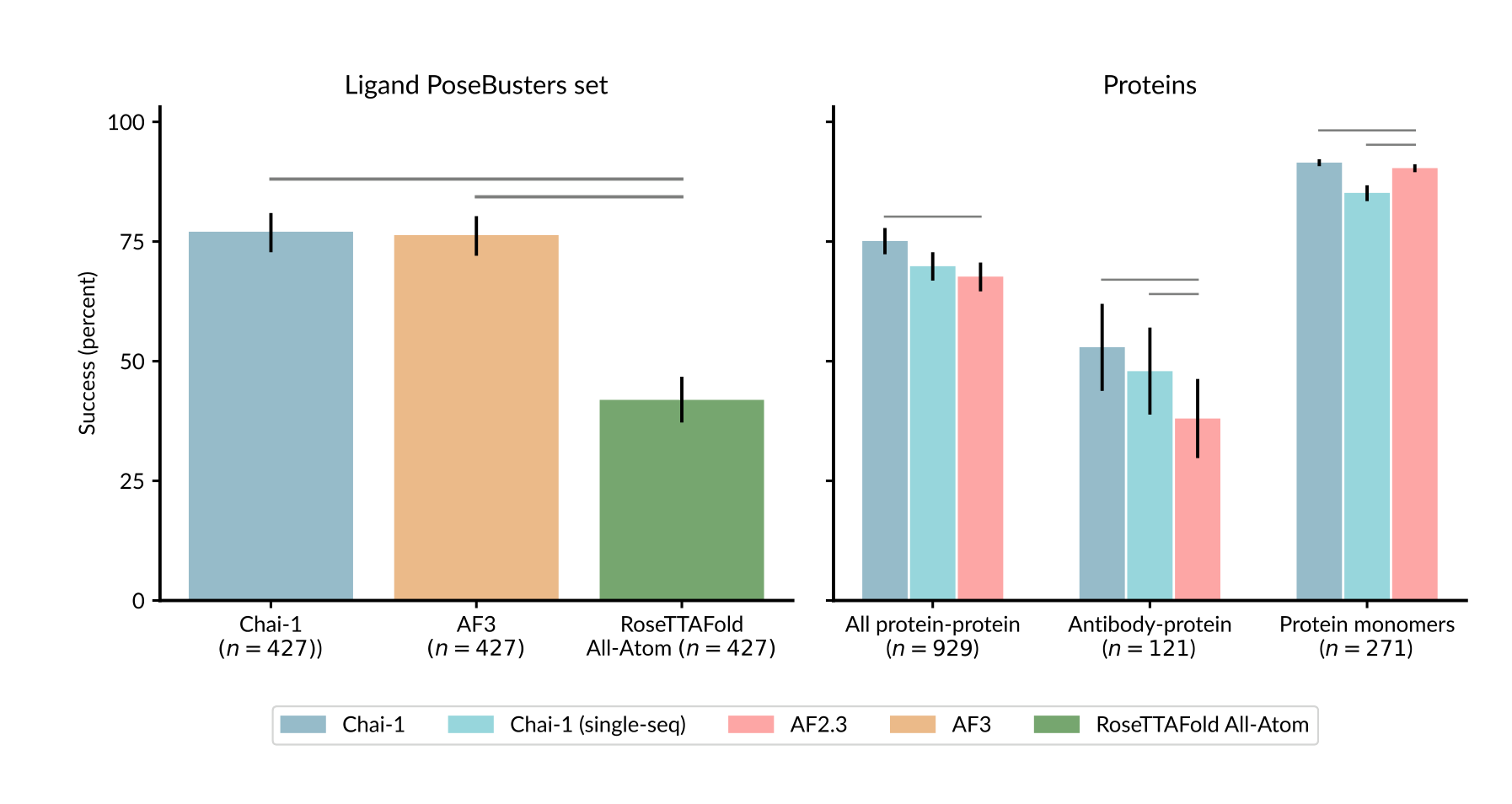
원문 Figure 2. PoseBusters, protein-protein, antibody-protein, protein monomer에서의 전반적 성능 비교.
논문에서 가장 눈에 띄는 숫자는 PoseBusters 결과다.
| 태스크 | Chai-1 | 비교 모델 |
|---|---|---|
| PoseBusters (ligand RMSD < 2 Å) | 77.05% | AF3 76.34%, RF2AA 42% |
| PoseBusters + docking conditioning | 81.20% | - |
이 결과가 중요한 이유는 두 가지다.
첫째, 리간드 포즈 예측에서 AF3와 거의 동급이라는 점이다.
둘째, apo 구조를 넣는 docking-style conditioning을 주면 81.2%까지 올라간다는 점이다.
여기서 읽어야 할 포인트는 “숫자 1~2% 차이”보다 prompting / conditioning이 실제로 먹힌다는 사실이다.
즉, Chai-1은 단순히 blind prediction만 잘하는 모델이 아니라, 추가 구조 정보가 들어오면 그걸 따라가는 모델이라는 뜻이다.
핵심 포인트
리간드 태스크에서 Chai-1의 강점은 “AF3와 비슷하다”보다, 실험 혹은 구조 힌트를 넣었을 때 더 좋아진다는 점에 있다.
가장 중요한 결과 2: multimer에서 AF2.3보다 강하고, single-seq도 실용적이다
논문 Table 2의 핵심 숫자만 뽑으면 아래와 같다.
| 태스크 | Chai-1 (with MSA) | Chai-1 (single-seq) | AlphaFold 2.3 multimer |
|---|---|---|---|
| Protein-protein | 0.751 | 0.698 | 0.677 |
| Antibody-protein | 0.529 | 0.479 | 0.380 |
| Protein monomers | 0.915 | 0.852 | 0.903 |
여기서 특히 중요한 해석은 다음과 같다.
Protein-protein
Chai-1은 protein-protein 인터페이스에서 AF2.3보다 분명히 앞선다.
더 흥미로운 점은 single-seq 모드(0.698) 가 AF2.3 with MSA (0.677) 와 비슷하거나 약간 더 높다는 것이다.
Antibody-protein
항체 관련 태스크에서는 격차가 더 크다.
Chai-1 with MSA는 0.529, AF2.3은 0.380이다.
그리고 single-seq도 0.479로 꽤 높다.
논문 저자들은 항체처럼 변이가 많고 진화 신호가 약한 경우, MSA가 주는 이득이 제한적일 수 있다고 해석한다. 그래서 Chai-1 single-seq 모드는 항체 설계나 면역계 단백질 탐색처럼 MSA가 약한 영역에서 특히 매력적이다.
Protein monomer
여기서는 균형 있게 봐야 한다.
with MSA 기준 Chai-1이 AF2.3보다 약간 낫지만, single-seq monomer는 AF2.3보다 약하다.
즉, 이 논문이 말하는 “single-seq 강점”은 모든 태스크를 다 이긴다는 뜻이 아니라,
특히 multimer / antibody / low-evolutionary-signal 영역에서 실용성이 높다는 뜻으로 읽어야 한다.
핵심 포인트
single-seq는 “MSA 없이도 어느 정도 된다” 수준이 아니라, 특정 태스크에서는 기존 MSA 기반 모델과 맞붙을 정도까지 왔다.
가장 중요한 결과 3: 제약조건은 실제로 구조 예측을 크게 끌어올린다
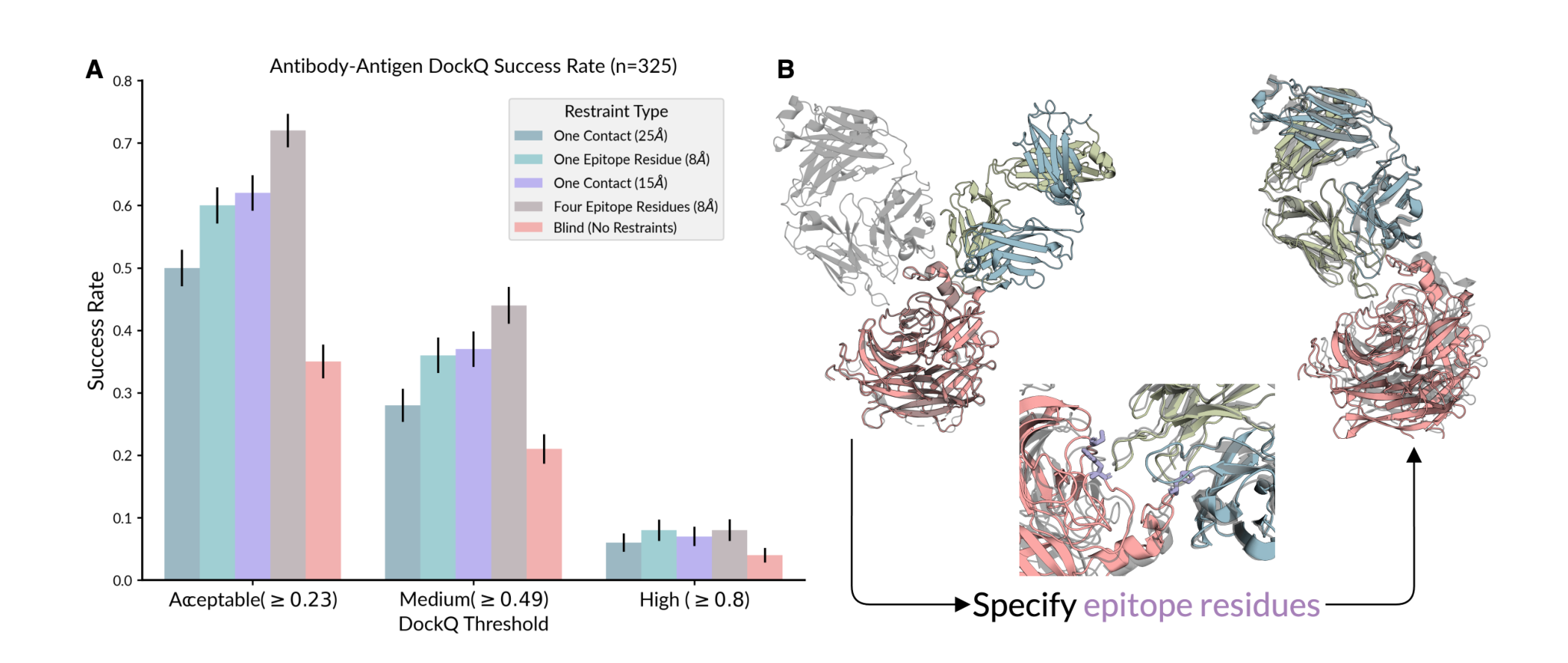
원문 Figure 4. 항체-항원 예측에서 contact / epitope 제약을 넣으면 성능이 뚜렷하게 개선된다.
이 그림은 Chai-1 논문 전체에서 가장 “실험 친화적”인 메시지를 담고 있다.
항체-항원 예측에서 baseline(Blind) acceptable DockQ 성공률은 35% 정도다.
그런데 제약을 넣으면 성능이 확 올라간다.
- 단일 거리 제약(one contact, 15 Å): acceptable 성공률이 35% → 57%
- epitope residue 4개 제공: 여러 품질 cutoff에서 baseline 대비 2배 이상 향상
물론 한계도 있다.
고품질(high quality, DockQ ≥ 0.8) 비율은 여전히 4~8% 수준으로 낮다. 즉, 제약조건이 도움은 되지만 항체-항원 구조 예측이 완전히 풀린 문제는 아니다.
그래도 이 결과는 매우 중요하다. 왜냐하면 실제 wet-lab에서는 완전한 구조보다 부분 제약 정보를 얻는 경우가 훨씬 많기 때문이다. Chai-1은 이 불완전한 실험 정보를 “프롬프트”처럼 구조 예측에 넣는 흐름을 보여준다.
핵심 포인트
이 논문의 가장 실무적인 메시지는 정확도 리더보드가 아니라, wet-lab 정보를 넣으면 어려운 complex 예측이 실제로 좋아진다는 점이다.
단백질 단량체와 핵산 결과는 어떻게 봐야 하나
논문은 monomer와 nucleic acid도 다룬다.
Protein monomer
- low-homology monomer set에서 Chai-1 with MSA는 AF2.3보다 약간 높다.
- CASP15 69개 타깃 평균 LDDT는 0.849, AF2.3은 0.843
- 특히 AF2.3이 어려워한 샘플에서는 개선폭이 더 컸다.
Nucleic acid
- Chai-1은 RNA MSA 없이 single-seq로 평가했다.
- 그럼에도 protein-RNA / protein-DNA / CASP15 RNA에서 RoseTTAFold2NA와 비슷한 수준을 보였다.
이 부분은 “압도적 승리”보다는 방향성이 중요하다.
즉, Chai-1은 핵산에서도 아예 못 쓰는 모델이 아니라, 아직 최적화 여지가 많지만 멀티모달 범용 모델로서 이미 usable한 수준에 도달했다는 의미다.
confidence score를 어느 정도 믿어도 되나
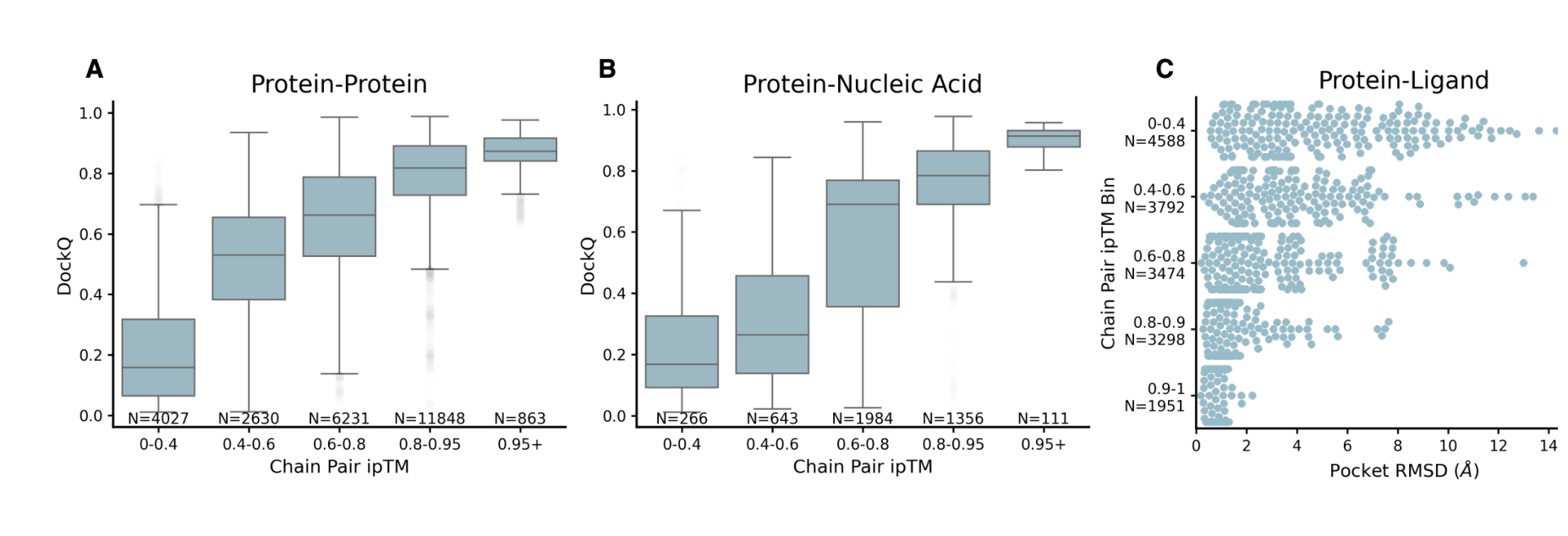
원문 Figure 5. ipTM confidence가 높을수록 protein-protein, protein-nucleic acid, protein-ligand에서 실제 품질이 좋아진다.
좋은 구조 모델은 “예측”만 하는 것이 아니라, 어떤 샘플을 믿어야 하는지도 알려줘야 한다.
이 논문에서 그 역할을 하는 것이 ipTM 기반 confidence다.
Figure 5를 보면 공통된 패턴이 있다.
- ipTM bin이 높아질수록
- DockQ는 올라가고
- ligand pocket RMSD는 내려간다
즉, Chai-1의 confidence score는 적어도 논문 평가셋에서는 랭킹 지표로 꽤 잘 작동한다.
이건 실무에서 매우 중요하다. 실제 사용자는 1개의 예측만 보는 것이 아니라 여러 샘플 중 무엇을 우선 검토할지 결정해야 하기 때문이다.
핵심 포인트
실전에서는 “최고 정확도”만큼이나 “무엇을 먼저 볼지”가 중요하다. Chai-1은 이 랭킹 문제에서도 쓸 만한 신호를 준다.
이 논문이 좋은 이유: 실패 사례를 숨기지 않는다

원문 Figure 3. 7Q2B 사례에서 Chai-1은 ground truth보다 더 깊은 ligand pose를 예측했지만, 실제 crystal 구조에는 DMS가 포켓 깊숙이 존재해 benchmark 자체에 해석 여지가 있음을 보여준다.
Figure 3은 개인적으로 이 논문의 가장 인상적인 장면 중 하나다.
저자들은 7Q2B 예시에서 Chai-1이 ligand를 포켓 더 안쪽에 배치해 “오답처럼 보이는” 예측을 내놓았다고 설명한다. 하지만 ground truth 구조에는 DMS(dimethyl sulfide) 가 포켓 깊숙이 들어가 있고, 이 분자가 crystallization aid라면 실제 ligand pose를 왜곡했을 가능성이 있다는 것이다.
이 사례가 말해주는 건 명확하다.
- 구조 예측 모델의 평가는 benchmark 숫자만으로 끝나지 않는다.
- crystal structure도 항상 절대적 진실은 아니다.
- 생성된 구조는 결국 사람이 직접 확인해야 할 후보다.
즉, Chai-1은 “벤치마크 점수 기계”가 아니라 실험 데이터와 함께 해석해야 하는 모델로 보는 편이 맞다.
실제 예시도 꽤 설득력 있다

원문 Figure 6. Zika protease + inhibitor, SARS-CoV-2 protease complex, PD-1 + cemiplimab 예시.
논문은 lab server 예시도 함께 보여준다.
- 7VLH: Zika virus NS2B-NS3 protease + small molecule inhibitor
- 7MB4: SARS-CoV-2 protease complex
- 7WVM: PD-1 + checkpoint inhibitor cemiplimab
이 예시의 의미는 “예쁜 그림” 이상이다.
저자들이 강조하고 싶은 것은 Chai-1이 단백질 하나를 접는 모델이 아니라, 치료적으로 의미 있는 복합체 구조를 다루는 모델이라는 점이다.
이 논문의 한계
좋은 요약은 장점만 정리하면 안 된다. 이 논문은 한계도 분명하다.
1) 상대적 orientation 실패
저자들은 개별 체인 자체는 맞게 예측해도, 체인들 사이의 상대 배치를 틀리는 경우가 있다고 인정한다.
복합체 예측에서 흔하지만, 실제 활용에서는 매우 치명적인 종류의 오류다.
2) modified residue에 민감
modified residue를 제거하거나 표준 아미노산으로 바꾸면 예측이 크게 달라질 수 있다고 한다.
즉, Chai-1은 modified residue를 “사소한 옵션”이 아니라 입력 의미 자체를 바꾸는 정보로 취급한다.
3) 항체-항원 고품질 예측은 아직 어렵다
restraining을 넣어도 high-quality 비율은 낮다.
항체-항원 docking은 여전히 어려운 문제라는 뜻이다.
4) 비교 가능성의 한계
논문도 분명히 적고 있듯이, protein-protein / antibody / protein-DNA / protein-RNA 수치는 AF3 논문의 평가셋과 정확히 같지 않다.
따라서 이 태스크들에서 AF3와 직접적인 우열 비교를 과하게 말하면 안 된다.
5) 아직 preprint다
이 논문은 peer review 전이다.
방향성과 결과는 매우 흥미롭지만, 최종 정리된 컨센서스로 받아들이기엔 이르다.
강조 메시지
메시지 1
“Chai-1은 AF3류 biomolecular structure prediction을 공개형으로 밀어낸 모델이다.”
메시지 2
“진짜 차별점은 정확도 1~2%가 아니라, wet-lab restraint를 구조 예측에 넣을 수 있다는 점이다.”
메시지 3
“single-sequence 모드가 생각보다 강해서, 특히 multimer와 antibody 작업에서 실용성이 높다.”
메시지 4
“항체-항원처럼 어려운 문제에서도 partial experimental signal이 있으면 성능이 크게 오른다.”
메시지 5
“벤치마크 점수만 보면 안 된다. crystal artifact와 alternative pose 가능성까지 같이 봐야 한다.”
최종 한 줄 평
Chai-1은 “또 하나의 구조 예측 모델”이 아니다.
이 논문은 멀티모달 분자 구조 예측 모델이 실험 정보와 어떻게 연결될 수 있는지를 꽤 설득력 있게 보여준다. 정확도 면에서도 강하지만, 더 중요한 것은 single-seq 실용성 + constraint conditioning + 공개 접근성이라는 조합이다.
참고 및 사용 주의
- 원문은 bioRxiv preprint이며, peer review 전 문서다.
- PDF 표지에는 CC BY-NC 4.0 라이선스가 표시되어 있다.
- 따라서 비상업적 블로그에서는 출처와 링크를 명확히 남기고 사용하는 방식이 일반적으로 더 안전하다.
- 다만 광고·수익화 블로그라면 commercial / non-commercial 해석이 운영 형태에 따라 달라질 수 있으므로, 원문 라이선스 조건을 한 번 더 확인하는 편이 좋다.
- 숫자 비교 중 일부는 논문 저자들이 직접 구성한 평가셋 기준이므로, 특히 AF3와의 비교는 태스크별로 직접 비교 가능성을 구분해서 써야 한다.



